


Síndrome del corazón izquierdo hipoplásico
Definición
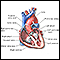
Es un padecimiento que ocurre cuando partes del lado izquierdo del corazón (válvula mitral, válvula aórtica, ventrículo izquierdo y aorta) no se desarrollan por completo. La afección es congénita (está presente al nacer).
Nombres alternativos
SCIH; Defecto congénito - corazón izquierdo hipoplásico; Cardiopatía cianótica - corazón izquierdo hipoplásico
Causas
El corazón izquierdo hipoplásico es un tipo poco común de enfermedad cardíaca congénita. Es más común en hombres que en mujeres.
Como sucede con la mayoría de los defectos cardíacos congénitos, no existe una causa conocida. Alrededor del 10% de los bebés que sufre este síndrome también tiene otros defectos de nacimiento. Esto también está asociado con algunas enfermedades genéticas tales como el síndrome de Turner, el síndrome de Jacobsen y la trisomía 13 y 18.
El problema se desarrolla antes del nacimiento cuando no hay un crecimiento suficiente del ventrículo izquierdo y otras estructuras, entre ellas:
- La aorta (el vaso sanguíneo que lleva sangre oxigenada desde el ventrículo izquierdo al cuerpo)
- Entrada y salida del ventrículo
- Válvulas mitral y aórtica
Esto lleva a un desarrollo incompleto o hipoplásico del ventrículo izquierdo y la aorta. En la mayoría de los casos, el ventrículo izquierdo y la aorta son mucho más pequeños de lo normal.
En los bebés con esta afección, el lado izquierdo del corazón no es capaz de enviar suficiente sangre al cuerpo. En consecuencia, el lado derecho del corazón debe mantener tanto la circulación pulmonar como la del cuerpo. El ventrículo derecho puede mantener la circulación tanto a los pulmones como al cuerpo por un tiempo, pero esta sobrecarga de trabajo lleva a que finalmente el lado derecho del corazón sufra insuficiencia.
La única posibilidad de supervivencia es una conexión entre el lado izquierdo y derecho del corazón o entre sus arterias y las arterias pulmonares (los vasos sanguíneos que llevan sangre a los pulmones). Los bebés nacen normalmente con dos de estas conexiones:
- El agujero oval (un orificio entre la aurícula derecha e izquierda)
- El conducto arterial (un pequeño vaso sanguíneo que conecta la aorta a la arteria pulmonar)
Ambas conexiones normalmente se cierran por sí solas unos pocos días después del nacimiento.
En los bebés con el síndrome del corazón izquierdo hipoplásico, la sangre que sale del lado derecho del corazón a través de la arteria pulmonar viaja a través del conducto arterial hasta la aorta. Esta es la única manera para que la sangre llegue al cuerpo. Si se deja que el conducto arterial se cierre en un bebé con el síndrome del corazón izquierdo hipoplásico, puede morir rápidamente debido a que no habrá ningún bombeo de sangre al cuerpo. A los bebés con este síndrome por lo regular se les comienza a dar un medicamento para mantener el conducto arterial abierto.
Debido a que hay poca o ninguna circulación del lado izquierdo del corazón, la sangre que retorna a este desde los pulmones necesita pasar a través del agujero oval o de una comunicación interauricular (un agujero que conecta las cámaras colectoras en los lados izquierdo y derecho del corazón) de nuevo hacia el lado derecho del corazón. Si no hay ningún agujero oval o si este es demasiado pequeño, el bebé podría morir. A los bebés con este problema se les abre el agujero entre las aurículas, ya sea con cirugía o usando una sonda delgada y flexible (cateterismo cardíaco).
Síntomas
Inicialmente, un recién nacido con corazón izquierdo hipoplásico puede parecer normal. Los síntomas pueden presentarse en las primeras horas de vida, aunque pueden demorar hasta algunos días en aparecer. Estos síntomas pueden ser:
- Color azulado de la piel (cianosis) debido a un nivel bajo de oxígeno en la sangre
- Manos y pies (extremidades) fríos
- Letargo
- Pulso débil
- Mala lactancia y alimentación
- Latidos cardíacos fuertes
- Respiración rápida
- Dificultad respiratoria
En los recién nacidos saludables, la piel morada en las manos y en los pies es una respuesta al frío (esta reacción se denomina cianosis periférica).
El color morado en el pecho o abdomen, en los labios y en la lengua es anormal (denominado cianosis central) y es un signo de que no hay suficiente oxígeno en la sangre. La cianosis central con frecuencia aumenta con el llanto.
Pruebas y exámenes
Un examen físico puede mostrar signos de insuficiencia cardíaca:
- Frecuencia cardíaca más rápida de lo normal
- Letargo
- Agrandamiento del hígado
- Respiración rápida
Además, el pulso puede estar muy débil en diversas localizaciones (muñeca, ingle y otros lugares). Al auscultar el tórax, a menudo (aunque no siempre) hay ruidos cardíacos anormales.
Los exámenes pueden incluir:
Tratamiento
Una vez que se hace el diagnóstico de corazón izquierdo hipoplásico, el bebé será llevado a la unidad de cuidados intensivos neonatales. Es posible que se necesite un respirador (ventilador) para ayudarle a respirar. Asimismo, se utiliza un medicamento llamado prostaglandina E1 para mantener la circulación de la sangre hacia el cuerpo, conservando el conducto arterial abierto.
Estas medidas no resuelven el problema. La afección requiere siempre cirugía.
La primera cirugía, llamada procedimiento de Norwood, se realiza en los primeros días de vida del bebé. Este procedimiento consiste en construir una nueva aorta:
- Usando la válvula y arteria pulmonares
- Conectando la aorta vieja hipoplásica y las arterias coronarias a la nueva aorta
- Quitando la pared entre las aurículas (tabique auricular)
- Haciendo una conexión artificial ya sea desde el ventrículo derecho o una arteria corporal hasta la arteria pulmonar para mantener la circulación a los pulmones (llamada derivación)
Se puede usar una variación del procedimiento de Norwood, llamado el procedimiento Sano. Con este procedimiento se crea un ventrículo derecho para la conexión de la arteria pulmonar.
Después de esto, el bebé se va a casa en la mayoría de los casos. El niño necesitará tomar medicamentos diarios y un cardiólogo pediátrico (especialista del corazón) deberá hacerle un seguimiento minucioso para determinar cuándo se debe llevar a cabo la segunda etapa de la cirugía.
La etapa II de la cirugía se denomina derivación de Glenn o procedimiento Hemifontan. También se le denomina derivación cavopulmonar. Este procedimiento conecta la mayor vena que lleva sangre sin oxígeno desde la mitad superior del cuerpo (vena cava superior) directamente a los vasos sanguíneos que van a los pulmones (arterias pulmonares) para obtener oxígeno. Esta cirugía casi siempre se realiza cuando el niño tiene entre 4 y 6 meses de edad.
Durante las etapas I y II, el niño aún puede lucir de color algo azul (cianótico).
La etapa III, el paso final, se denomina procedimiento de Fontan. El resto de las venas que llevan sangre azul desde el cuerpo (vena cava inferior) se conecta directamente a los vasos sanguíneos que van a los pulmones. El ventrículo derecho ahora sirve solo como la cámara de bombeo para el cuerpo (ya no más los pulmones y el cuerpo). Esta cirugía generalmente se lleva a cabo cuando el bebé tiene entre 18 meses y 4 años de edad. Después de este último paso, el bebé ya no se pondrá cianótico y tendrá un nivel normal de oxígeno en la sangre.
Algunas personas pueden necesitar más cirugías cuando llegan a los 20 o 30 años en caso de que presenten arritmias difíciles de controlar u otras complicaciones del procedimiento de Fontan.
Algunos médicos consideran el trasplante de corazón como una alternativa a la cirugía de los 3 pasos. Pero hay pocos corazones donados disponibles para niños pequeños.
Expectativas (pronóstico)
Sin tratamiento, el síndrome del corazón izquierdo hipoplásico es mortal. Las tasas de supervivencia para la reparación por etapas siguen aumentando a medida que mejoran la técnica quirúrgica y el tratamiento posoperatorio. La supervivencia después de la primera etapa es de más del 75%. Los niños que sobreviven a su primer año tienen una muy buena posibilidad de supervivencia a largo plazo.
El desenlace clínico del niño después de la cirugía depende del tamaño y funcionamiento del ventrículo derecho.
Posibles complicaciones
Las complicaciones incluyen:
- Obstrucción de la derivación artificial
- Coágulos sanguíneos que pueden llevar a accidente cerebrovascular o embolia pulmonar
- Diarrea prolongada (crónica) (a partir de una enfermedad llamada enteropatía causante de pérdida de proteínas)
- Líquido en el abdomen (ascitis) y en los pulmones (derrame pleural)
- Insuficiencia cardíaca
- Ritmos cardíacos rápidos e irregulares (arritmias)
- Accidentes cerebrovasculares y otras complicaciones del sistema nervioso
- Deterioro neurológico
- Muerte súbita
Cuándo contactar a un profesional médico
Llame a su proveedor de atención médica de inmediato si su bebé:
- Come menos (disminución en la alimentación)
- Tiene la piel azulada (cianótico)
- Tiene nuevos cambios en los patrones respiratorios
Prevención
No se conoce prevención para el síndrome del corazón izquierdo hipoplásico y, como sucede con muchas enfermedades congénitas, sus causas son inciertas y no ha sido asociado a ninguna enfermedad ni comportamiento de la madre.
Las mujeres que planean quedar embarazadas deben vacunarse contra la rubéola si aún no son inmunes. La infección por rubéola en una mujer embarazada puede causar enfermedad cardíaca congénita.
Las mujeres embarazadas deben recibir una buena atención prenatal:
- Evitar el alcohol y las drogas ilegales durante el embarazo.
- Informarle a su proveedor que están en embarazo antes de comenzar a tomar cualquier medicamento nuevo.
- Realizarse un examen de sangre al inicio del embarazo para saber si son inmunes a la rubéola. Si no son inmunes, evitar cualquier exposición a la rubéola y vacunarse inmediatamente después del parto.
- Las mujeres embarazadas que tengan diabetes deben tratar de tener un buen control de su nivel de azúcar en la sangre.
Referencias
Bernstein D. Cyanotic congenital heart disease: evaluation of the critically ill neonate with cyanosis and respiratory distress. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 478.
Kliegman RM, St. Geme JW, Blum NJ, et al. Cyanotic congenital heart disease: lesions associated with increased pulmonary blood flow. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 480.
Valente AM, Dorfman AL, Babu-Narayan SV, Kreiger EV. Congenital heart disease in the adolescent and adult. In: Libby P, Bonow RO, Mann DL, Tomaselli GF, Bhatt DL, Solomon SD, eds. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 12th ed. Philadelphia, PA: Elsevier; 2022:chap 82.
Well A, Fraser CD. Congenital heart disease. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery. 21st ed. St Louis, MO: Elsevier; 2022:chap 59.
Versión en inglés revisada por: Thomas S. Metkus MD, PhD, Associate Professor of Medicine and Surgery, Johns Hopkins University School of Medicine, Baltimore, MD. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
Traducción y localización realizada por: HolaDoctor
 Todos los derechos son reservados
Todos los derechos son reservados