

Alport syndrome
Definition
Alport syndrome is a rare inherited disorder that damages the tiny blood vessels in the kidneys. It can also cause hearing loss and eye problems.
Alternative Names
Hereditary nephritis; Hematuria - nephropathy - deafness; Hemorrhagic familial nephritis; Hereditary deafness and nephropathy
Causes
Alport syndrome is an inherited form of kidney inflammation (nephritis). It is caused by a variant in a gene for a protein in the connective tissue, called collagen.
The disorder is rare. There are three genetic types:
- X-linked Alport syndrome (XLAS) -- This is the most common type. The disease is more severe in males than in females.
- Autosomal recessive Alport syndrome (ARAS) -- Males and females have equally severe disease.
- Autosomal dominant Alport syndrome (ADAS) -- This is the rarest type. Males and females have equally severe disease.
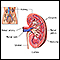
With all types of Alport syndrome, the kidneys are affected. The tiny blood vessels in the glomeruli of the kidneys are damaged. The glomeruli filter blood to make urine and remove waste products from the blood.
At first, there are no symptoms. Over time, as the glomeruli are more and more damaged, kidney function is lost and waste products and fluids build up in the body. The condition can progress to end-stage renal disease (ESRD) at an early age, between adolescence and age 40. At this point, dialysis or a kidney transplant is needed.
Symptoms
The main symptom of Alport syndrome is blood in the urine that is often not visible and only detected under a microscope. This occurs from a young age. As the disease progresses, other symptoms may appear.
KIDNEYS
Other symptoms of kidney problems include:
- Abnormal urine color
- Blood in the urine that is visible during a cold or flu or exercise
- Flank pain
- High blood pressure
- Swelling throughout the body (edema)
- Fatigue
- Poor appetite
- Excessive thirst
EARS
Over time, Alport syndrome also leads to hearing loss in both ears. By the early teens, it is common in males with XLAS, though in females, hearing loss is not as common and happens when they're adults. With ARAS, boys and girls have hearing loss during childhood. With ADAS, it occurs later in life.
Hearing loss usually occurs before kidney failure.
EYES
Alport syndrome also leads to eye problems in those with XLAS and ARAS, including:
- Abnormal shape of the lens (anterior lenticonus), which can lead to a slow decline in vision as well as cataracts.
- Corneal erosion in which there is damage to the outer layer of the covering of the eyeball, leading to pain, itching, redness of the eye, or blurred vision.
- Abnormal coloring of the retina, a condition called dot-and-fleck retinopathy. It doesn't cause vision problems, but can help diagnose Alport syndrome.
- Macular hole in which there is thinning or a break in the macula. The macula is a part of the retina that makes central vision sharper and more detailed. A macular hole causes blurred or distorted central vision.
Eye problems are uncommon in people with ADAS.
Exams and Tests
The health care provider will examine you and ask about your symptoms.
The following tests may be done:
- BUN and serum creatinine
- Complete blood count
- Renal biopsy
- Urinalysis
If your provider suspects you have Alport syndrome, you will also likely have vision and hearing tests.
Treatment
The goals of treatment include monitoring and controlling the disease and treating the symptoms.
Your provider may recommend any of the following:
- A diet that limits salt, fluids, and potassium
- Medicines to control high blood pressure
Kidney disease is managed by:
- Taking medicines to slow kidney damage
- A diet that limits salt, fluids, and protein
Hearing loss can be managed with hearing aids. Eye problems are treated as needed. For example, an abnormal lens due to lenticonus or cataracts can be replaced.
Genetic counseling may be recommended because the disorder is inherited.
Support Groups
More information and support for people with Alport syndrome and their families can be found at:
- Alport Syndrome Foundation -- alportsyndrome.org/for-patients/patient-resources/
- National Kidney Foundation -- www.kidney.org/atoz/content/alport
- National Organization for Rare Disorders -- rarediseases.org/rare-diseases/alport-syndrome
Outlook (Prognosis)
The outlook depends on the type of Alport syndrome, biological sex, and age.
For both males and females, kidney function will worsen over time. In males with XLAS, this may lead to kidney failure at an earlier age. Most men with XLAS will experience kidney failure by age 60. Women with XLAS may or may not have kidney problems, but the risk increases with age.
Both males and females with ARAS will develop kidney failure by early adulthood.
ADAS progresses slowly in both males and females, and kidney failure may not occur until later in life.
As kidneys fail, dialysis or a kidney transplant will be needed. Kidney transplant is often very successful in people with Alport syndrome.
When to Contact a Medical Professional
Contact your provider for an appointment if:
- You have symptoms of Alport syndrome.
- You have a family history of Alport syndrome and you are planning to get pregnant.
- Your urine output decreases or stops or you see blood in your urine (this may be a symptom of chronic kidney disease).
Prevention
Awareness of risk factors, such as a family history of the disorder, may allow the condition to be detected early.
References
Gregory MC. Alport syndrome and related disorders. In: Gilbert SJ, ed. National Kidney Foundation's Primer on Kidney Diseases. 8th ed. Philadelphia, PA: Elsevier; 2023:chap 41.
Radhakrishnan J, Appel GB, D'Agati VD. Secondary glomerular disease. In: Yu ASL, Chertow GM, Luyckx VA, Marsden PA, Skorecki K, Taal MW, eds. Brenner and Rector's The Kidney. 11th ed. Philadelphia, PA: Elsevier; 2020:chap 32.
Radhakrishnan J, Stokes MB. Glomerular disorders and nephrotic syndromes. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 107.
Rheault MN, Kashtan CE. Alport syndrome and other familial glomerular syndromes. In: Johnson RJ, Floege J, Tonelli M, eds. Comprehensive Clinical Nephrology. 7th ed. Philadelphia, PA: Elsevier; 2024:chap 48.
|
Review Date:
4/1/2025 Reviewed By: Walead Latif, MD, Nephrologist and Clinical Associate Professor, Rutgers Medical School, Newark, NJ. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. |
© 1997- A.D.A.M., a business unit of Ebix, Inc. Any duplication or distribution of the information contained herein is strictly prohibited.
 All rights reserved.
All rights reserved.